Conceptos clave:
- Principios de la termodinámicaü
- Termoquímica: ley de Hessü
- Entropíaü
- Energía de Gibbs y espontaneidad de los procesos químicosü
- Concepto de equilibrio químicoü
- Constantes de equilibrioü
- Relación energía de Gibbs-constante de equilibrioü
- Ley de Le Chatelierü
- Producto de solubilidadü
- Efecto del ión comúnü
Objetivos:
- Enunciar los principios de la termodinámicaü
- Aprender a calcular entalpías de reacción a partir de entalpías de formación, entalpías de enlace o entalpías de reacción (Ley de Hess)ü
- Predecir la espontaneidad de una reacción químicaü
- Resolver problemas de equilibrio químico homogéneo y heterogéneoü
- Resolver problemas de solubilidadü
- Predecir el efecto de variaciones de presión, volumen, temperatura y concentración sobre la posición de equilibrio de una reacciónü
Sistemas, variables de estado, funciones de estado, ecuaciones de estado.
Para la termodinámica, el Universo se divide en dos: el sistema bajo estudio y el resto del Universo. Las relaciones entre ambos permiten clasificar los sistemas del siguiente modo:
¿Intercambia
materia?
|
¿Intercambia
energía?
|
|
Abierto
|
Sí
|
Sí
|
Cerrado
|
No
|
Sí
|
Adiabático
|
Sí
|
No
|
Aislado
|
No
|
No
|
Las magnitudes que definen una situación o estado del sistema se denominan variables de estado, como Q, W, p, V, T o n. A veces se puede encontrar una ecuación que las relaciona, dichas ecuaciones se llaman ecuaciones de estado, por ejemplo, pV=nRT. Finalmente, dentro de las variables de estado, existe un grupo que tiene la particularidad de que su variación sólo depende de los estados final e inicial, y no de cómo se pasa de uno a otro, estas magnitudes son las funciones de estado, y algunas de ellas son la energía interna, la entalpía, la entropía y la energía de Gibbs.
En lo sucesivo, usaremos el siguiente criterio de signo para el calor (Q) y el trabajo (W):

Los cuatro principios de la termodinámica.
Principio cero. Si dos sistemas están en contacto, y entre ellos no fluye energía, entonces están en equilibrio térmico y comparten la misma temperatura.

Primer principio. El calor suministrado a un sistema, y el trabajo realizado sobre él, incrementan su energía interna.
Q+W=DU
Segundo principio. La entropía del Universo aumenta en cualquier proceso irreversible y permanece constante en los reversibles.
DSU>=0
Tercer principio. La entropía de un elemento químico puro y cristalino a 0 K es 0.
Primer principio de la termodinámica. Termoquímica.
El trabajo en los sistemas químicos está restringido a la expansión o contracción de los gases involucrados. Consideremos entonces cierto gas confinado en un émbolo cilíndrico de sección S con un pistón.

Si aplicamos una fuerza F y desplazamos el pistón una distancia d, entonces habremos hecho un trabajo sobre el gas (positivo) igual a
W=F·d
dado que p=F/S, podemos reescribir la expresión anterior:
W=F·d=P·S·d=P·V
puesto que la sección del émbolo por el desplazamiento nos da un volumen; dicho volumen es exactamente la diferencia ente el volumen inicial del gas y el volumen final, es decir, V=Vi-Vf=-DV, pues los incrementos se calculan exactamente al revés, así que finalmente obtenemos:
W=F·d=P·S·d=P·V=-P·DV
Si utilizamos esta expresión para el trabajo en la expresión del primer principio.
Consideremos ahora las condiciones habituales de trabajo en el laboratorio:
DU=Q+W=Q-P·DV
Consideremos ahora las condiciones habituales de trabajo en el laboratorio:
- si V=cte., entonces Qv=DU
- si P=cte, entonces Qp=DU+P·DV=(Uf-Ui)+P·(Vf-Vi)=(Uf+P·Vf)-(Ui+P·Vi)=Hf-Hi=DH
La entalpía y la energía interna de un proceso están relacionadas entre sí:
DH=DU+P·DV=DU+DnRT
donde Dn es la variación del número de moles de gas en todo el proceso, dado que los sólidos y líquidos no se tienen en cuenta. En procesos en los que no se produce tal variación, la energía interna y la entalpía coinciden numéricamente.
Como ya se ha dicho, la entalpía es una función de estado, y como tal, su variación sólo depende de cuáles son el estado inicial y final y no de cómo se ha producido el paso de uno a otro. Esto es similar a lo que ocurre con la energía potencial en Física: uno puede medir variaciones de la misma pero no valores absolutos. Pues bien, en Termodinámica, se adopta la misma solución que en Física: asignar un estado de referencia, un "cero", respecto del cual se miden luego las entalpías. El estado de referencia para las entalpías es el siguiente:
"La entalpía de los elementos químicos en condiciones estándar (CCSS: 1 atm, 25 ºC) en su estado natural es 0"
O sea, que la entalpía del Fe sólido en CCSS es cero, no así la del hierro fundido. O por ejemplo, la entalpía del yodo sólido (I2)en CCSS es cero, pero no así la del vapor de yodo.
Tomando ese estado de referencia, se pueden obtener las entalpías de formación de los compuestos químicos. Se define entalpía de formación como la energía puesta en juego a presión constante cuando se forma un mol de compuesto a partir de los elementos en estado estándar.
Una vez que esas entalpías están TABULADAS, podemos emplearlas para calcular la entalpía de cualqueir reacción teniendo en cuenta la propiedad fundamental de las funciones de estado, aplicada a las entalpías:
DHºR=(SHºf)p-(SHºf)r
es decir, que la entalpía de cualquier reacción se puede calcular restando las entalpías de formación de los productos y las de los reactivos. Las entalpías de reacción se pueden incorporar a los cálculos estequiométricos convencionales.
Una reacción muy frecuente es la de combustión, y es posible incluso encontrar tablas de entalpías de combustión. Ésta se define como la energía liberada a presión constante cuando se quema un mol de compuesto en estado estándar.
Igualmente se pueden encontrar entalpías de disolución, de vaporización, etc.
Una segunda opción para determinar entalpías de reacción es la de aplicar el mismo principio pero implicando a otras reacciones, una combinación de las cuáles es capaz de llevarnos a la reacción problema. Esto se denomina principio de aditividad de las reacciones químicas o ley de Hess. En pocas palabras dice que si puedes combinar linealmente varias reacciones químicas entre sí, la entalpía de la reacción resultante será igual a la misma combinación lineal de las entalpías de las reacciones combinadas.
Finalmente, aún tenemos una tercera posibilidad, y supone emplear las entalpías de enlace. Se denomina entalpía de enlace a la energía necesaria para romper un mol de enlaces químicos en fase gaseosa, y es un valor que se determina promediando las energías del enlace en cuestión en una serie de compuestos químicos comparables (Enlace a tabla de entalpías de enlace). Al ser el resultado de un promedio, los valores de entalpía calculados a partir de entalpías de enlace son necesariamente aproximados y peores que los determinados a partir de entalpías de formación.
DHºR=(SHºenlaces rotos)-(SHºenlaces formados)
Dado que estas entalpías son un promedio, obviamente el resultado que se obtiene de ellas es siempre menos preciso que el que se puede obtener de las entalpías de formación.
Segundo y tercer principios de la termodinámica. Espontaneidad de los procesos químicos.
Los procesos en la naturaleza se pueden dividir en dos categorías: reversibles e irreversibles. Los procesos reversibles son procesos ideales en los que para ir del estado inicial al final se pasa por infinitos estados intermedios de equilibrio, descritos cada uno de ellos de forma continua por la ecuación de estado del sistema. Dado que es necesario un tiempo para el establecimiento del equilibrio, estos procesos se llaman a veces cuasiestáticos. En la práctica, los procesos naturales son todos irreversibles, es decir suceden sin pasar por infinitos estados de equilibrio, por ejemplo una compresión brusca de una gas. ¿Por qué distinguir estos dos tipos de procesos si el primero no se da de facto? Porque aunque eso sea así, las funciones de estado van a variar lo mismo independientemente de cómo se produzca el proceso, y podremos calcular su variación mediante las ecuaciones de estado, aunque en realidad la cosa suceda de otra forma.
Pues bien, el segundo principio establece que
La entropía es una función de estado difícil de comprender. Para procesos a temperatura constante, como los cambios de estado, la variación de entropía es el calor reversible puesto en juego dividido por la temperatura absoluta:
Así, para la fusión del hielo, el calor puesto en juego es positivo, puesto que es absorbido, y la variación de entropía es positiva, mientras que para la congelación sería negativa.
Hay diversas aproximaciones al concepto de entropía: la más común es considerarla una medida del grado de desorden de un sistema, de modo que cuanto más desordenado está, más entropía posee. Otra interpretación la asemeja al discurrir del tiempo, pues como éste, no hace más que aumentar: el Universo es una máquina de generar entropía. Sin embargo, asociándola al orden, sí posee una característica que la distingue a de la entalpía, y es que es fácil pensar en un estado de máximo orden al que asignar entropía mínima, o sea 0. Esto es lo que establece el tercer principio de la termodinámica:
Al final, la entropía y el segundo principio lo que hacen es constatar que en la naturaleza, los sistemas tienden de manera natural al desorden, y que mantenerlos ordenados siempre supone consumir energía. Si unimos esta tendencia natural a la otra que está latente en el primer principio, esto es, que los sistemas tienen a situaciones de mínima energía, tendremos los dos "motores" que impulsan los procesos naturales en general y químicos en particular. En termodinámica, existe una función de estado que aúna ambas tendencias, es la energía de Gibbs y se define como
De ese modo, es fácil darse cuenta que un proceso exotérmico (evoluciona a menor energía) en el que aumenta el desorden (entropía positiva) debe tener DG negativa, y será natural que ocurra, como por ejemplo pueden se la combustión del papel. Sin embargo, uno endotérmico y que produce un estado más ordenado tendrá DG positiva y no ocurrirá, como por ejemplo que a temperatura ambiente un vaso de agua se congele.
Estas observaciones permiten establecer la condición de espontaneidad de los procesos químicos en función del signo de DG. Así diremos que:
Empleando la definición de la energía de Gibbs es posible desgranar estas condiciones un poco más:

Pues bien, el segundo principio establece que
"la entropía del Universo aumenta en los procesos irreversibles y se mantiene constante en los reversibles."
DSU>=0
La entropía es una función de estado difícil de comprender. Para procesos a temperatura constante, como los cambios de estado, la variación de entropía es el calor reversible puesto en juego dividido por la temperatura absoluta:
DS=Qrev/T
Así, para la fusión del hielo, el calor puesto en juego es positivo, puesto que es absorbido, y la variación de entropía es positiva, mientras que para la congelación sería negativa.
Hay diversas aproximaciones al concepto de entropía: la más común es considerarla una medida del grado de desorden de un sistema, de modo que cuanto más desordenado está, más entropía posee. Otra interpretación la asemeja al discurrir del tiempo, pues como éste, no hace más que aumentar: el Universo es una máquina de generar entropía. Sin embargo, asociándola al orden, sí posee una característica que la distingue a de la entalpía, y es que es fácil pensar en un estado de máximo orden al que asignar entropía mínima, o sea 0. Esto es lo que establece el tercer principio de la termodinámica:
"La entropía de un elemento químico puro y cristalino a 0 K es 0."
Al final, la entropía y el segundo principio lo que hacen es constatar que en la naturaleza, los sistemas tienden de manera natural al desorden, y que mantenerlos ordenados siempre supone consumir energía. Si unimos esta tendencia natural a la otra que está latente en el primer principio, esto es, que los sistemas tienen a situaciones de mínima energía, tendremos los dos "motores" que impulsan los procesos naturales en general y químicos en particular. En termodinámica, existe una función de estado que aúna ambas tendencias, es la energía de Gibbs y se define como
DG=DH-T·DS
De ese modo, es fácil darse cuenta que un proceso exotérmico (evoluciona a menor energía) en el que aumenta el desorden (entropía positiva) debe tener DG negativa, y será natural que ocurra, como por ejemplo pueden se la combustión del papel. Sin embargo, uno endotérmico y que produce un estado más ordenado tendrá DG positiva y no ocurrirá, como por ejemplo que a temperatura ambiente un vaso de agua se congele.
Estas observaciones permiten establecer la condición de espontaneidad de los procesos químicos en función del signo de DG. Así diremos que:
- un proceso con DG negativa será espontáneo
- con DG positiva será no espontáneo, y
- si DG vale 0 entonces nos encontramos en situación de equilibrio termodinámico que veremos un poco más adelante.
Empleando la definición de la energía de Gibbs es posible desgranar estas condiciones un poco más:
- Procesos exotérmicos, DH<0
- Si DS>0, siempre será espontáneo
- Si DS<0, dependerá de la temperatura,
- si T es alta, será no espontáneo
- si T es baja, será espontáneo
- Procesos endotérmicos, DH>0
- Si DS<0, siempre será no espontáneo
- Si DS>0, dependerá de la temperatura,
- si T es alta, será espontáneo
- si T es baja, será no espontáneo
Problemas termodinámica v3 from Víctor Manuel Jiménez Suárez
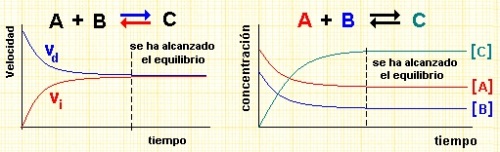


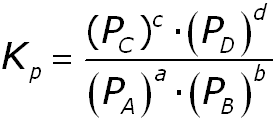

Equilibrio químico
Sea un proceso elemental genérico A + B = C + D, y supongamos que inicialmente sólo tenemos reactivos. Conforme vaya produciéndose la reacción, la concentración de reactivos bajará e irá aumentando la de los productos, de modo que comenzará a ser posible que ocurra la reacción inversa C + D = A + B. Conforme pase el tiempo, la velocidad de la reacción directa disminuirá al ir disminuyendo la concentración de los reactivos, y la de la inversa aumentará, al hacerlo la concentración de productos. En un determinado momento, ambas reacciones tendrán la misma velocidad, y los productos se formarán a la misma velocidad que se recombinan en los reactivos originales, resultando que las concentraciones permanecen constante. Es ese momento diremos que la reacción está en equilibrio químico (DG=0), y como queda demostrado por el modo en que se ha descrito, es una situación dinámica, pues las reacciones no se han detenido, sino que han "congelado" una determinada situación.
Estas reacciones se indican usando la doble flecha, como en la figura. Para ver cómo se establece el equilibrio a nivel molecular haz clic en este ENLACE.
Constantes de equilibrio
Como hemos visto arriba, en el equilibrio, las velocidades directa e inversa se igualan, por lo tanto:
vd=kd·[A]·[B]
vi=ki·[C]·[D]
cuando lleguemos al equilibrio, vd=vi, y entonces kd·[A]·[B]=ki·[C]·[D],
agrupando constantes:
donde Kc es la constante de equilibrio. Recordemos que las constantes de velocidad solo dependen de la temperatura; por consiguiente, la constante de equilibrio, que es un cociente de dos constantes de velocidad, también depende solamente de la temperatura.
Para una reacción genérica:
a A + b B = c C + d D,
Esta expresión se conoce como ley de acción de masas de Guldberg y Waage.
La constante de equilibrio también puede expresarse en términos de presión parcial de los gases implicados (cuando la reacción sólo implique a gases). En ese caso, la expresión quedaría:
Kp y Kc están relacionadas, dado que pV=nRT, y entonces, p=(n/V)RT, y n/V es la concentración molar o molaridad del gas considerado:
Independientemente de la expresión que usemos, si el valor de la constante de equilibrio es mucho mayor que 1, significa que hay mucho más producto que reactivo y se dice que el equilibrio está desplazado hacia los productos. Si por el contrario, es mucho menor que 1, ocurre al revés y el equilibrio está desplazado hacia los reactivos.
Dado un conjunto de concentraciones de sustancias iniciales, podemos saber cómo va a evolucionar el sistema calculando el denominado cociente de reacción Q, que es la misma expresión que la constante de equilibrio, pero con las concentraciones en cualquier instante de la reacción, no necesariamente cuando se ha alcanzado el equilibrio.
- Si Q=Kc, el sistema está en equilibrio
- Si Q>Kc, sobran productos y faltan reactivos, y el sistema evolucionará hacia la izquierda
- Si Q<Kc, ocurrirá al revés y evolucionará hacia la derecha
Relación Kc - Energía de Gibbs
La variación de energía de Gibbs para condiciones no estándar está relacionada con la variación de energía de Gibbs estándar y el cociente de reacción de la siguiente forma:

A partir de ahí, y considerando que la energía de Gibbs en equilibrio será 0 y Q será igual a K, se puede obtener

Cabe indicar que esa constante no es ni Kp ni Kc, sino la constante termodinámica, en la que las sustancias disueltas se expresan en concentración molar y los gases en atmósferas. Cuando se trata de equilibrios homogéneos entre gases, K=Kp, y cuando se trata de equilibrios homogéneos en disolución, K=Kc.
@VicMJim Thermodynamic Equilibrium Constant (from old copy of Ebbing&Gammon): pic.twitter.com/ppSXfHOjTI
— Andy Napper (@QuantumDot2) March 9, 2015
Grado de disociación
Este es un concepto usado con bastante frecuencia en los problemas de equilibrio. Se define como el cociente entre la cantidad de sustancia disociada y la cantidad inicial, pudiéndose a veces indicar como tanto por ciento:

Equilibrios heterogéneos
Hasta ahora, todos los equilibrios tratados consistían de una única fase homogénea, bien fuera gaseosa o bien en disolución. Se llaman equilibrios heterogéneos aquellos que presentan más de una fase, por ejemplo: un sólido y gases, o una disolución y un sólido. La única diferencia con los equilibrios homogéneos es que los sólidos y líquidos puros no aparecerán en la constante de equilibrio, tan sólo los gases o las sustancias en disolución. Por ejemplo, para la reacción: CaCO3(s) = CaO(s) + CO2(g), Kp=P(CO2)
Ley de Le Chatelier
Básicamente, la ley de Le Chatelier dice que el equilibro químico se opone a cualquier cambio que se haga sobre la posición de equilibrio. Eso se consigue desplazando el equilibrio hacia uno de los lados.
Un equilibrio se puede alterar de diferentes formas:
- Cambiando la temperatura. Este es el único caso en que la constante de equilibrio cambia. Si el equilibrio es exotérmico y aumentamos la temperatura, tratará de compensarlo, absorbiendo energía desplazándose en sentido endotérmico, o sea en este caso, hacia los reactivos. Por o tanto, la constante disminuye. Si el equilibrio es endotérmico ocurriría al revés.
- Cambiando las cantidades de alguna de las sustancias involucradas. Eso se puede lograr añadiendo más disolución o su presión parcial. Si por ejemplo se aumenta, el equilibrio se desplazará hacia el lado contrario al que está, y al revés si se disminuye.
- Actuando sobre la presión total. Eso se puede conseguir, por ejemplo, cambiando el volumen, y es un cambio que afecta solo a los gases. Si se aumenta la presión, el equilibrio se desplaza hacia el lado en que haya menos moles de gas. En los equilibrios donde no varían los moles de gas, no se altera la posición de equilibrio.
| Click to Run |
Problemas de equilibrio químico v2 from Víctor Manuel Jiménez Suárez

Solublidad. Producto de solubilidad.
Se llama solubilidad a la cantidad máxima de un soluto que se pueden disolver en 100 g de un disolvente. Representa la concentración de la disolución saturada a una determinada temperatura.
Los equilibrios de precipitación son aquellos en los que se representa el proceso de disolución de una sustancia, generalmente poco soluble, por lo que en el equilibrio tenemos un sólido en equilibrio con sus iones en disolución. Por ejemplo:
AgCl(s) D Ag+(aq) + Cl-(aq)
PbI2(s) D Pb2+(aq) + 2 I-(aq)
Las constantes de estos equilibrios se denominan producto de solubilidad (Ks, Kps ó Ps), y siguen las reglas vistas con anterioridad:
Ps(AgCl)=[Ag+(aq)]·[Cl-(aq)]
Ps(PbI2)=[Pb2+(aq)]·[I-(aq)]2
Si llamamos s a la solubilidad, se puede relacionar con el producto de solubilidad. Por ejemplo, si hay disueltos s mol/L de yoduro de plomo(II), habrá en el equilibrio s mol/L de iones plomo(II) y 2s mol/L de iones yoduro, y de ahí:
Ps(PbI2)=[Pb2+(aq)]·[I-(aq)]2=s·(2s)2=4s3;
s=(Ps/4)1/3
Efecto del ión común.
Llamamos así al descenso de la solubilidad de una sustancia cuando junto a ella, se disuelve otra, usualmente muy soluble, que comparte con ella un ión. Se puede considerar una consecuencia del principio de Le Chatelier, aplicado al caso concreto de los equilibrios de precipitación: al haber una fuente "externa" de iones (productos) el equilibrio de precipitación se desplaza hacia los reactivos, y disminuye así la solubilidad.
Formación de precipitados.
Que una sustancia precipite o no dependerá de que se supere o no su producto de solubilidad, de modo que basta con sustituir en la fórmula del mismo las concentraciones iniciales y comparar el resultado con el dado. Si lo supera, entonces precipitará.
Redisolución de precipitados.
Un precipitado puede redisolverse actuando sobre el equilibrio de precipitación, de modo que se desplace hacia los productos (los iones). Esto se puede conseguir de muy diversas formas, por ejemplo, los haluros de plata se pueden redisover añadiendo amoniaco, que formará con los iones plata un ión complejo que consigue desplazar el equilibrio en el sentido de la disolución del sólido; o los hidróxidos metálicos, que se pueden redisolver bajando el pH añadiendo algún ácido, o el yoduro de plomo(II), que se puede redisolver calentando la disolución.
que largo
ResponderEliminarque largo
ResponderEliminara sus ordenes mi comandante
ResponderEliminarsuuuuuuuuuuu
ResponderEliminar